
本文章由GeneDireX技術支援提供
生命科學是包括對所有生物體像是:微生物、動物、植物等,進行研究的一門學科;近年來得力於分子生物學和生物技術上的快速發展,讓專業研究人員和一般民眾都對生命科學研究有新的應用和認識,從而改變我們的生活方式。而分子生物學更是生命科學領域裡一門專注於以分子為單位進行的研究學科,藉由微觀細胞中基因體及DNA、RNA、蛋白質等分子的變化,或利用特定的技術來建構轉殖基因以研究基因功能與特性。
在分子生物學實驗室中,將特定的基因序列插入指定載體(vector)中並與此載體接合(ligation)成為一個質體(plasmid),接著再將該質體轉型(transformation)到大腸桿菌細胞中,使其可以被複製或被表現蛋白;而這就是分子生物學技術中最常見也最常用的實驗技術:分子選殖或分子克隆(molecular cloning)。藉由這個技術,我們不僅可以以經濟又快速的方法獲得大量的特定基因片段,也可以任意的剪輯基因序列,增加或減少特定基因序列來達成操控基因的目的。
然而,並非所有轉型到大腸桿菌細胞中含有目標基因的質體都有成功轉移入菌落,因此會需要針對單一菌落檢查是否帶有轉入目標基因質體。最直接的做法是挑選單一菌落進行微量培養、質體DNA抽取、限制酶作用和電泳確認;但是這樣做相當耗時且耗費實驗室資源。因此對於檢測的方法,會是用不同於一般勞動力密集的方法;而是更省時有用的像是:藍/白篩、條件性致死基因表達、菌落PCR。簡單的來說,藍/白篩即是利用beta-galactosidase,可以分解 X-gal (5-bromo-4-chloro-3-indolyl-β-D-galactopyranoside),形成藍色沉澱。
因此帶有目標基因成功接入的菌落呈現白色,反之則為藍色;或是放入致死基因,只有成功接入目標基因的菌落的得以存活;反之失敗的菌落就會死亡。上述的實驗原理都是利用目標基因插入位置可以破壞原本帶有的可表達beta-galactosidase基因或是致死基因,成功插入者便可以被藉此挑選出來。另外一類則是菌落PCR;與一般PCR原理步驟相同,利用特定的引子序列(primer)進行基因序列放大反應,配合電泳確認產物大小,挑選出有成功帶有目標基因的質體。
進行菌落PCR時,其引子序列設計可以是兩股都位在插入片段特異性的引子、載體特異性的引子、或者一股在插入片段一股在載體上(圖一);而不同的設計策略對於後續的判讀也會不同(圖二)。

圖一
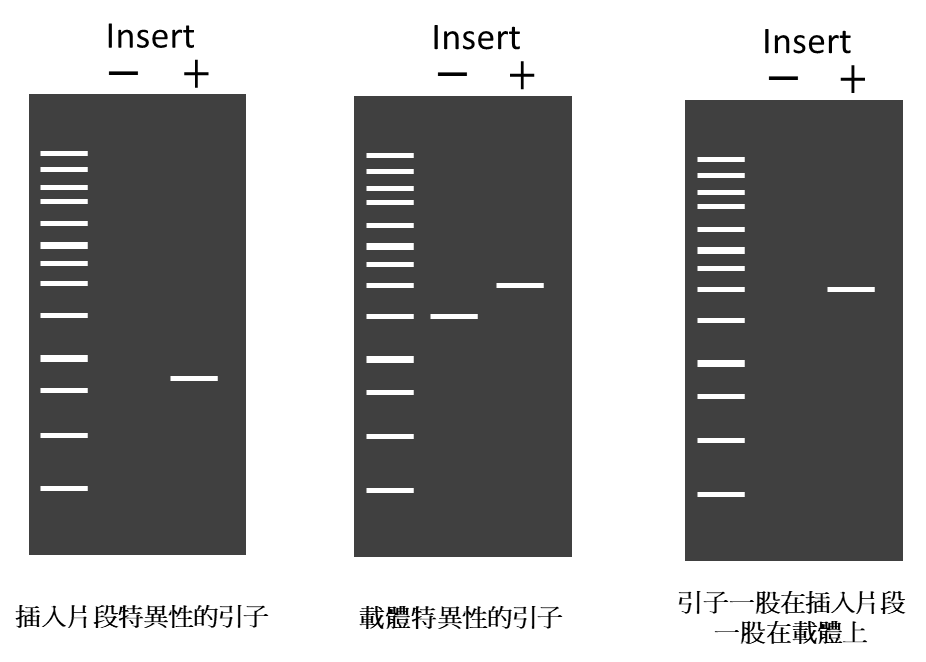
圖二
菌落PCR雖然可以方便我們快速篩選出可能帶有成功接入目標基因的載體;但是仍有造成錯誤的機會發生。
1998年Frances M. Sladek 發表在BioTechniques的一篇文章(BioTechniques 24:580-582, April 1998)提出,當我們將質體藉由轉型作用送入大腸桿菌細胞後,殘留在溶液內或是大腸桿菌細胞表面的質體會一起被塗佈到LB-agat plate上。當我們在挑選菌落進行PCR反應時,這樣的基因片段就會因此被放大,進而造成誤判。
推測其原因在於 PCR反應的靈敏度非常好,可以偵測到0.4pg的質體DNA。實驗室進行類似的實驗設計,我們將一段序列D200接入vector中,並將後續引子序列設計在D200序列上;菌落PCR結果顯示有挑中3個(lane 1-3);但是隨機在LB agar plate,無菌落處的PCR結果也有微弱訊號(lane 4 and 5);lane 6和7則是NTC (non template control)(圖三);在將此3個進行微量培養、質體DNA抽取和相同的PCR反應則沒有呈現出預期的結果(圖四,lane 2-4;lane 1 D200, positive control;lane 5-10, negative control)。
這樣的實驗數據也就說明菌落PCR雖然方便、快速;但是還是需要後續的實驗來確認是否有成功挑選到帶有目標基因的菌落。
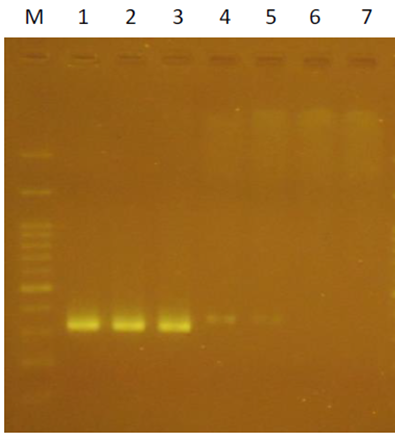
圖三
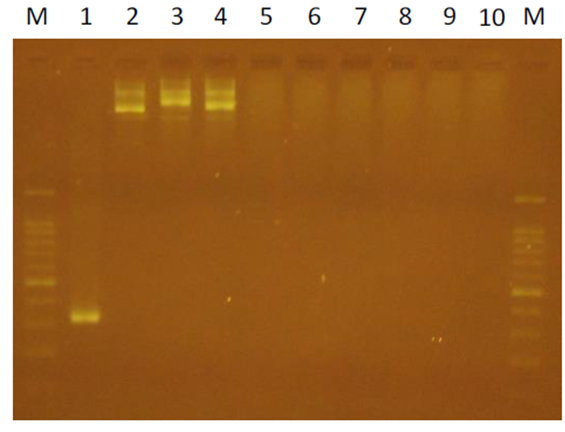
圖四